Chapter XXIII. Production Of Acetone By Means Other Than Fermentation
Description
This section is from the book "Distillation Principles And Processes", by Sydney Young. Also available from Amazon: Distillation Principles And Processes.
Chapter XXIII. Production Of Acetone By Means Other Than Fermentation
Manufacture of Acetone from Acetate of Lime, etc. - Acetone is produced commercially by the dry distillation of various acetates, calcium acetate (commercial grey acetate of lime) or barium acetate being generally used. The single acetate is usually employed, although mixtures of two acetates have been recommended (F.P. 439732/1911). Grey calcium acetate usually contains about 80 per cent calcium acetate, the remaining 20 per cent consisting of water and various impurities, including small quantities of calcium formate and propionate as well as salts of other organic acids.
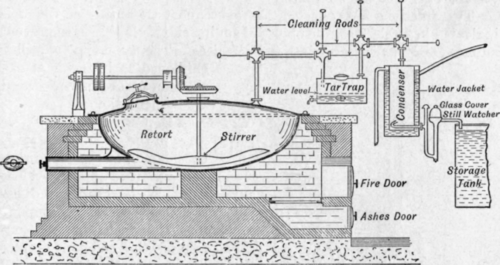
Fig. 93.
Calcium acetate when heated at a temperature of about 380° C. decomposes, giving acetone and a residue of calcium carbonate, but at the same time the accompanying calcium salts present as impurities also react and acetaldehyde and various higher ketones are formed together with the condensation products. In addition other impurities (such as dumasin) and tar-like bodies are formed.
The distillation of calcium acetate is carried out generally in a shallow circular retort taking a charge of from 300 lb. to 700 lb. Fig. 93 gives, an outline drawing (kindly supplied by Mr. W. T. Thomson, F.I.C.) of the usual type of retort employed.
The heating of the retort is carried out by direct fire. Stirring gear is provided and the outlet tubes are fitted with rods passing through stuffing joints and having an iron disc fitted on the end so that the pipes may be cleaned of dust, tar, etc., at intervals during the distillation. After the retort has been charged the stirring gear is started, and heating is begun moderately at first, as acetone and water are given off at a moderate temperature. The temperature is gradually increased up to 380° C, when the decomposition of the acetate proceeds briskly. Steam is blown through at the end to sweep through the last portion of the distillate. The vapour from the retort passes first through a tank in which the greater part of the dust and tar are retained. Acetone and water vapour mixed with volatile impurities pass on, and are condensed in a worm and collected in a galvanised tank. Towards the end of the distillation the contents of the dust and tar tank are heated by a steam coil to drive out any acetone which may have condensed with the tar.
The distillate is diluted with water till the specific gravity reaches 0.960. The liquid is then well stirred and allowed to stand for 12 hours. The tar, higher ketones, etc., which rise to the top are run off by means of a tap to an adjoining tank. To the crude liquor containing acetone is then added 3.5 lb. of sodium hydrate per 1000 gallons of liquid. The liquid is then well stirred and transferred to a rectifying still. The addition of the sodium hydrate solution prevents volatile acids from passing over into the distillate.
Two fractions are collected, the subsequent treatment of which is indicated in Table 95. The object of adding sulphuric acid is to prevent amines from passing over into the distillate. The same type of still is employed as in the final distillation of acetone produced by fermentation. «
The presence of alkali (free lime) in the calcium acetate is considered to be a disadvantage if a high yield is wanted. Becker1 introduces a stream of carbon dioxide into the heated vessel containing the calcium acetate to prevent the formation of free lime. For the preparation of ketones by the decomposition by heat of certain calcium salts in a current of carbon dioxide, there is an earlier reference to that of Becker. In 1891, S. Young2 prepared dibenzyl ketone in good yield by heating dry calcium phenylacetate in a current of carbon dioxide. A short description of the apparatus (Fig. 94) and method as employed on the laboratory scale is given. The calcium salt dried at 130° was placed in a wide
1 Zts. Ang. Chem., 1907, 20, 206. 2 Trans. Chem. Soc, 1891, 59, 621.

Fig. 94.
Table 95 cylindrical glass bulb sealed to a narrower tube. The bulb was heated by the vapour of sulphur boiling in a flanged iron vessel provided with a heavy iron cover. The narrow tube already referred to was provided with an india-rubber stopper, through which a T-tube passed. A piece of barometer tubing enlarged conically at its lower end passed through the T-tube and the narrow tube. It was attached to the T-tube by a cork. The barometer tubing was bent as shown in Fig. 94, after the T-tube and the cork had been placed in position. It is possible that a bath containing a mixture of equal molecular proportions of potassium and sodium nitrates might with advantage be substituted for the sulphur bath. The limit of temperature for the distillation should be 370 to 446°.

The overheating due to direct contact of the acetate with the hot walls of the retort has been largely overcome by the employment of a retort made by F. H. Meyer of Hanover. Fig. 95 shows a retort of this type. Trays of acetate are placed upon a perforated truck which is wheeled bodily into the retort. By this method a spent charge is readily withdrawn and a fresh charge inserted with little loss of heat, and the avoidance of the dusty operation of withdrawing spent lime.

A method somewhat related to the above consists in passing a continuous current of pyroligneous acid over a heated acetate capable of forming acetone.1
Conversion Of Acetic Acid To Acetone In Presence Of A Catalyst
When the vapours of acetic acid are passed into air-tight vessels containing some porous material saturated with lime or baryta, a good conversion to acetone is brought about.2
The methods developed at the Shawinigan Falls, Canada, for the preparation of acetone from synthetic acetic acid have recently been described by Matheson.1 In the experimental installation three tubes (four inch) 6 feet in length were employed. They were heated electrically by means of a resistance winding and the acid was vaporised before passing into the tube. The catalyst consisted of hydrated lime mixed with a small amount of magnesia deposited on pumice stone. The yield obtained was about .95 per cent of the theoretical. When the process was developed to large scale experiment, initial difficulties were encountered, due to the difficulty of heat transference. Fig. 96 is a diagram of the acetone process as worked at the Shawinigan Falls. The conversion vessels consist of steel tubes, 13 feet in length, 12 inches in diameter with centrally heated core. They were filled with cast - iron balls on which the catalyst was placed by immersing the balls in a paste of the catalyst and drying them in the tube by the passage of a current of air. Seventy-two of these tubes were installed and this installation was sufficient for the production of ten tons of acetone per day. Fig. 97 shows the details of one of the conversion tubes. The mixed vapours of acetone, water, and unconverted acetic acid were combined from 24 of these tubes to the one main leading to alkali scrubbers maintained at 98° C. by the heat from the gases ; the acetone and water vapours which passed the scrubbers were condensed to give a 20 per cent aqueous solution of acetone. This mixture was rectified in an ordinary continuous acetone still. The optimum temperature for conversion was 485° C. Each tube was supplied with three thermo-couples having leads to a central switch-board and the temperature was maintained constant by installing the resistance windings in parallel so that any of the parallel windings could be cut out as the temperature fluctuated. Iron had a detrimental action on the conversion, but this difficultywas eliminated by coating the walls of the tube with the catalyst itself. Copper is the best metal to employ for the tubes, but its cost is very high. After every fourteen days it is necessary to renew the catalyst by recoating the balls.
1 J. Soc. Chem. Ind,, 1906, 25, 634; 1907, 26, 1002 ; 1908, 27, 277.
2 J. Soc. Chem. Ind., 1899, 18, 128, 828. Bauschlicker, D.R.P. 81914.

1 Canadian Chem. J., Aug. 1919.

Fig. 97.
The average efficiency of the process is about 85 per cent, and the acetone produced passes the strictest commercial specifications.
Acetone From Wood Spirit
The process of removing acetone from methyl alcohol on the manufacturing scale is described by Mariller, La Distillation fractionnee, 1917, p. 402.
The purification of the acetone so obtained has, however, not until now been accomplished on the large scale. Attempts to do so were made in Canada by Messrs. Barbet and Fils and Co. of Paris, but they say that they failed to obtain a finished product containing more than 92 per cent of acetone. Certain impurities, particularly methyl alcohol, remained in the acetone and could neither be removed by rectification nor by chemical treatment.
Methyl alcohol and acetone give an azeotropic binary mixture boiling at 55.95° C. and containing 86.5 per cent of acetone in the distilled liquid. While the presence of azeotropic mixtures of alcohol, methyl alcohol, and such a substance as methyl acetate, has not been definitely proved, yet in practice such a ternary mixture boils at nearly constant temperature. In the distillation of wood spirit there is the possibility of the formation of various azeotropic binary and probably of ternary mixtures, and for this reason it is practically impossible to obtain a pure acetone by distillation and fractionation alone.
Acetone and carbon disulphide give an azeotropic mixture boiling at 39.25° and containing 34 per cent by weight of acetone in the mixture. On this fact has been based a process for the purification of acetone by Duclaux and Lavzenberg.1 To the acetone is added 1.7 times its volume of carbon disulphide. The distillate between 38-40° is extracted with water and the aqueous solution distilled. The pure acetone is collected at 56.1° to 56.3°.
When methyl alcohol is present, as in the mixture from wood spirit, the process must be modified since methyl alcohol and carbon disulphide give an azeotropic mixture boiling at 37.5°. The azeo-tropic mixtures are left over potassium carbonate for some time before extraction with water, and copper sulphate is added to the aqueous solution extract before its fractionation. In this way it is claimed that most of the methyl alcohol is removed.
Acetone From Another Source
Another method which has been suggested for the preparation of acetone depends upon the action of a reducing agent or amine upon a monobasic acid or ester.2
1 Bull. Soc. Chem., 1920 (IV.), 779-782, Cf. also J. Chem. Soc. (A), 1920, 118, 1, 818.
2 P. de la Fresnaye and E. Cadorat de la Gabiniere, F.P. 451374/1912, abst. J. Soc. Chem. Ind., 1913, 32, 625.
Continue to:
My Books

