Chapter Seven. Quanta Associated With Running Water
Description
This section is from the book "Vivaxis, The Spiral Of Life", by Frances Nixon. Also available from Amazon: Vivaxis, The Spiral Of Life.
Chapter Seven. Quanta Associated With Running Water
We have seen how energies from a body of moving water underground can be used for polarizing. Both water and atomic alignment combine to play an active role in our biology. Our body is said to consist of approximately 80 per cent water. The atomic character of water is well explained by the following:
Water is vital to all earthly life. It is not surprising that this substance plays a role in virtually all cell activities. The atoms of HoO are not arranged in a linear pattern. They form a V, the oxygen being located at the pointed end. The two hydrogens are attached to the oxygen because they are all wrestling for the same tiny particles, called electrons. The oxygen is usually getting the best of the tussle, so the electrons spend more time in its neighbourhood. Since electrons have negative electrical charges, the oxygen is somewhat negative, so the hydrogens have no choice but to be positive. We see, then, that the water molecule is lop-sided as to electrical charge. This is why it is said to be polar - having two different kinds of poles.*
Moving bodies of water underground appear to create their own Vivaxis. A plausible explanation might be as follows (figure 7):
*The Human Machine, Harry Moody.
The colliding particles in the water create a kinetic energy which stimulates the moving molecules of water to align their electron spins with the earth's magnetism and with each other. The energies aligning to north and south in horizontal planes are pulled in to a common axis; where they meet, a Vivaxis is formed. At this point where horizontal waves of energy meet they change direction into a vertical plane - the energy bundles, quanta, carrying the pattern of radiation energy associated with the area of its Vivaxis. The local fields of energy on the ground tend to become pulled down toward the water's Vivaxis - the age-old principle of a weaker current being attracted by a stronger - a balancing of nature's energies.
A brief example will illustrate how strong these forces are.
"X" refers to a person standing on ground where there is a water Vivaxis vertically beneath him. This point of ground we will call W.V.
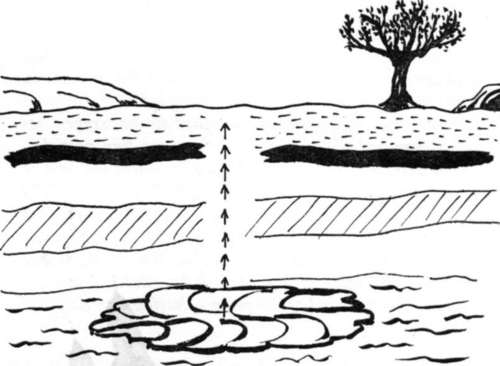
Figure 7. Concept of underground water Vivaxis.
Test (a)
"X" positions himself over point W.V. facing out of direct alignment to his own Vivaxis. He stays there for ten seconds before moving. This is done with his eyes shut at first and then, open.
Results of Test (a)
With his eyes open, radiation associated with the streams of quanta travelling from the water's Vivaxis had been absorbed by X's thyroid. This is detected in his thyroid after he moves from point W.V.
With his eyes closed, no radiation was absorbed, indicating that light is the carrier wave.
Test (b)
X stands over point W.V., but this time he positions into the channel to his own Vivaxis. He remains for ten seconds. This is also done with eyes open and then closed.
Result of Test (b)
Wave tests of his thyroid do not show any radiation associated with point W.V. The thyroid shows only radiation associated with X's own Vivaxis.
Irrespective of the eyes being open or shut, no radiation from the water Vivaxis is absorbed. Here is one of the first of many examples of how alignment to one's own Vivaxis has the added strength and ability to influence or cancel foreign radiation waves.
Vivaxis of water can be accurately pin-pointed. Why does a willow wand pull down? We have established two known reasons. (1) It pulls down to a Vivaxis of running water under the ground when the diviner is not in alignment to his own Vivaxis, and (2), it pulls down also to a person's own Vivaxis, providing it is located at a geographically lower altitude than where he is currently standing. This occurs when he is positioned head and body erect and faced directly aligned to his own Vivaxis - frontwards, sideways, and backwards. When his back is toward his Vivaxis, the wand usually pulls in toward himself, and down; if frontwards, away from himself and down. If his shoulders are sideways to his Vivaxis, the wand pulls away and down, but when he turns the opposite way, it pulls in and down.
Ignorance of this fact of our biology and physics has undoubtedly been responsible for many poorly located wells. According to the driller's theory, water tables can always be found in porous sandstone and limestone. The yardstick of water divining is to accurately determine the depth of water and is not to be confused with the depth of the diviner's Vivaxis.
All of the above applies only to those who are free from the interfering energy fields of X-rays.
The precision with which a polarized magnetic wave other than your own can be traced to its Vivaxis is illustrated here.
I have located in all four wells. Each time prior to drilling I have accurately calculated the different depths at which each water table would be found. The last well was drilled on our property at Clam Bay and is detailed in the signed report. The results were signed and confirmed by Mr. E. G. Peet, the man in charge, working on the job for Pacific Water Drillers.
To those who are not familiar with modern drilling methods - as the drill spins downward, dry powdered rock is blown off by air pressure. When a water table is reached, water spurts up. Often it is necessary to drill through one or two tables to find a sufficient supply. As the drilling proceeds, the powdered rock gets drier and drier until the next water table is struck. In between the calculated levels of this well there was nothing but dry rock.
Prior to drilling this well I handed Mr. Peet a slip of paper giving the three depth measurements that each table of water would be located. I explained I wanted this for scientific records. He voiced his apprehension, and if I had been a sensitive soul, or one with less valid reason for confidence, I would have smarted from his ridicule.
Instead, I challenged him to put up his money and insisted we make it small as the odds were unfair to him. I not only won the bet, but he quite humbly said, "I didn't think this was possible. You have taught me a lesson I never will forget. I thought guessing that accurately would be absolutely impossible."
I use this example to show that water detecting is a real science. It illustrates that if the wave-transmitter carefully keeps his wave pattern weaker by positioning out of his own Vivaxis channel, he can then trace a polarized wave to its birthplace. This principle has to be carefully adhered to in all of our fact-finding.
I have often heard diviners mention that they detect an underground stream crossing at right angles. If their own Vivaxis was located at an elevation below their testing ground, the chances are they were merely pulling down towards the horizontal leading to their own Vivaxis. Under these circumstances, when they are faced aligned toward the direction of their own Vivaxis, a forked instrument in their hands will pull down. When they are faced at right angles in their wave channel, it will also pull down. The unawareness of this fact of their body's energy circuit can account for the fact that many say depths cannot be reliably determined. Part of the time they are locating their own Vivaxis and the height they are located above it.
Continue to:
My Books

