Fatty Acid Metabolism Of Clostridium Kluyveri. Continued
Description
This section is from the book "The Scientific Contributions Of The Ben May Laboratory For Cancer Research", by The University of Chicago. Also available from Amazon: The Scientific Contributions Of The Ben May Laboratory For Cancer Research.
Fatty Acid Metabolism Of Clostridium Kluyveri. Continued
Secondly, the role of acetoacetate must be considered. Again, this compound itself cannot be a true intermediate since it cannot be reduced to butyrate under hydrogen, under conditions suitable for synthesis of butyrate from acetyl phosphate and acetate. Although these extracts contain an enzyme capable of the phosphoroclastic splitting of acetoacetate to acetate and acetyl phosphate, this reaction does not occur at pH values optimal tor butyrate oxidation. These data suggest that the accumulation of acetoacetate when butyrate is oxidized in the absence of inorganic phosphate is due to a side reaction, in which the true intermediate is converted by a relatively irreversible step to acetoacetate.
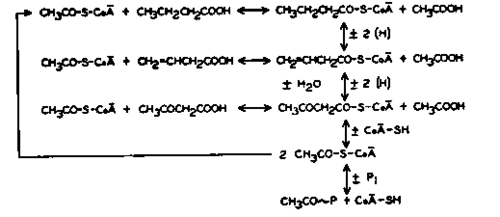
Fig. 3.
Finally, there is the fact that butyrate oxidation proceeds at a significant rate in the absence of inorganic phosphate. This finding suggests that inorganic phosphate participates only in the final formation of acetyl phosphate and acetate by cleavage of the oxidized product at the acetoacetate level.
To account for these facts, a scheme was suggested for butyrate oxidation which is essentially that shown in Fig. 3. According to this hypothesis, the first step in butyrate oxidation is a reduction of butyrate with CoA. The CoA itself may require a preliminary activation by reaction with acetyl phosphate. This is definitely indicated by the fact that acetyl phosphate is required for butyrate oxidation by dialyzed enzyme preparations. From the work of Lynen, Reichert, and Rueff (48) on the nature of the activated acetate-GoA complex, it is reasonable to postulate a butyryl thioester of CoA as the true intermediate. This compound then undergoes successive oxidation reactions to the vinylacetyl-CoA and acetoacetyl-CoA levels. Free vinylacetate and acetoacetate enter and leave the reaction scheme by formation or splitting off from the respective CoA-compounds. It is necessary to assume that vinyl acetyl-CoA is relatively easily formed in this system, while free acetoacetate is not readily activated.
In the presence of inorganic phosphate, the acetoacetyl-CoA compound breaks down with the net production of one mole of acetyl phosphate and one mole of acetate for each mole of butyrate oxidized. In view of the findings of Stadtman, Doudoroff, and Lipmann (58) on the mechanism of acetoacetate formation with a mammalian enzyme, and the results obtained by Lieberman and Barker (3) in their study of the breakdown of β-ketovalerate in C. kluyveri extracts, it is to be expected that this breakdown is quite complex, and may well involve the formation of two moles of acetyl-CoA, with subsequent utilization of one mole to activate further amounts of butyrate in cyclic fashion.
While the proposed scheme has the merit of accounting for the puzzling lack of free intermediates in the butyrate oxidation scheme, it is obviously not the only explanation which could be put forward. On the other hand the recent direct evidence for the formation of fatty acid-CoA compounds put forward by Kornberg (31) and by Drysdale (14) strengthens the suggested mechanism considerably. Furthermore, the scheme accounts for the lack of randomization of isotopic carbon in acetoacetate recovered from experiments with carboxyl-labeled butyrate, since a splitting off of acetoacetate as a 4-carbon unit is to be expected if this mechanism were correct. The possibility that such a direct splitting off of acetoacetate may account in part for the complex factors governing the isotope distribution in acetoacetate formed during fatty acid oxidation in mammalian tissues has already been discussed.
It is of importance to consider to what extent fatty acid oxidation in C. kluyveri resembles analogous processes in mammalian tissues. On the whole, it may be concluded that the similarities are much more important than the observed differences. In both cases, the process proceeds by a fundamentally similar pattern of β-oxida-tion, with removal of 2-carbon units which are identifiable with acetyl coenzyme A. In both cases, the oxidative reaction appears to be initiated by activation of the fatty acid molecule at the expense of a high-energy priming compound, either acetyl phosphate in C. kluyveri extracts, or ATP in the mammalian enzymes. Of course, due attention must be paid to the fact that animal tissues lack phos-photransacetylase, and therefore acetyl phosphate will neither be formed from nor activate fatty acid oxidation in enzymes from liver or kidney. Furthermore, the general pattern of fatty acid synthesis in C. kluyveri and in mammalian tissues appears to be fundamentally similar. Barker, Kamen, and Bornstein (4) showed that fatty acid synthesis in the microorganism proceeds by the condensation of 2-carbon units, and that the fatty acid chains are elongated by the addition of the 2-carbon unit to the carboxyl rather than to the omega end of the preexisting fatty acid. Work by Anker (1) and by Zabin (66) has demonstrated an identical pathway in mammals.
Setting aside, then, obvious differences in the nature of the electron transfer system, and allowing for the important differences brought about by the presence of the enzyme phosphotransacetylase in the bacterial extracts, it would appear reasonable to assume that fatty acid oxidation and synthesis in C. kluyveri may serve as a valuable model for similar processes in animal tissues. In this connection, there are two differences in the respective enzyme systems worth noting. Fatty acid synthesis in C. kluyveri appears to be a direct reversal of the oxidative mechanism. If this were also true of animal tissues, then isolated mitochondria, which carry out vigorous oxidation of fatty acids, should also be expected to carry out fatty acid synthesis, if proper conditions could be provided. It would be, of course, advantageous in many ways to work with cell-free preparations rather than with intact slices in studying the effect of insulin, for example, on the process of fatty acid synthesis. While early experiments of Bloch met with little success in this direction, recently Brady and Gurin (7) have reported some encouraging results in studying fatty acid synthesis in homogenates. Secondly, the high degree of specificity of the bacterial enzyme for vinyl acetate must be contrasted with the ready oxidation of other 4-carbon acids at the same level of oxidation reported for animal enzymes. Thus crotonate and isocrotonate are completely inert in C. kluyveri extracts, while these compounds, as well as vinyl acetate, are attacked by liver and kidney. Lipmann and Perlmann (46) have reported results on the metabolism of crotonate in kidney slices and homogenates which are remarkably suggestive of the behavior of vinyl acetate in the bacterial enzyme system. It is possible that separate oxidative pathways exist for vinyl acetate and crotonate in mammalian tissue, or alternatively, crotonate may first undergo transformation to vinyl acetate or vinyl acetyl CoA by an enzyme presumably missing in the bacterial extracts.
If the scheme for fatty acid oxidation in C. kluyveri given in Fig. 3 has some validity for the analogous processes in animal tissues, then one puzzling feature of work with animal fatty acid oxidases is at once explained, namely, the failure to demonstrate intermediates between the long-chain fatty acids and the C2 level. Such intermediates would not be found, simply because they do not exist as free carboxylic acids. The mechanism of splitting off acetyl-CoA units by the addition of a second molecule of CoA-SH at the B position gives rise to a fatty acid shortened by 2-carbon atoms, and already activated for further oxidation. Isotope tracer work with C. kluyveri has shown that the free ^-oxidized compounds are not in isotopic equilibrium with the activated CoA conjugates, which are the true intermediates.
References
1. Anker, H. S-, J. Biol. Chem., 194, 177 (1952).
2. Atchley, W. A., J. Biol. Chem., 176, 123 (1948).
3. Barker, H. A., in Phosphorus Metabolism. I (W. D. McElroy & B. Glass, eds.), Johns Hopkins Press, Baltimore (1951).
4. Barker, H. A., Kamen. M. D., and Bornstein, B. T., Proc. Natl. Acad. Sci., 31, 373 (1945).
5. Bloch, K., Physiol. Rev., 27, 574 (1947).
6. Bloch, K., and Rittenberg, D., J. Biol. Chem., 159, 45 (1945).
7. Brady, R. O, and Gurin, S., Federation Proc, 11, 190 (1952).
8. Breusch, F. L., Science, 97, 490 (1943).
9. Buchanan, J. M., Sakami, W., and Gurin, S.,J. Biol. Chem., 169, 411 (1947).
10. Chaikoff, I. L., Goldman, D. S., Brown, G. W., Dauben, W. G-, and Gee, M., J. Biol. Chem., 190, 229 (1951).
11. Crandall, D. I., Brady, R. C, and Gurin, S., J. Biol. Chem., 181, 845 (1949).
12. Crandall, D. I., and Gurin, S., J. Biol. Chem., 181, 829 (1949).
13. Cross, R. J., Taggart, J. V., Covo, G. A., and Green, D. E., J. Biol. Chem., 177, 655 (1949).
14. Drysdale, G. R., Federation Proc, 11, 204 (1952).
15. Friedkin, M. E., and Lehninger, A. L., J. Biol. Chem., 178, 611 (1949).
16. Geyer, R. P., Cunningham, M., and Pendergast, J., J. Biol. Chem., 185, 461 (1950) .
17. Geyer, R. P., Cunningham, M., and Pendergast, J., J. Biol. Chem., 188, 185 (1951) .
18. Geyer, R. P., Matthews, L. W., and Stare, F. J., J. Biol. Chem., 180, 1037 (1950).
19. Grafflin, A. L., and Green, D. E., J. Biol. Chem., 176, 95 (1948).
20. Green, D. E., Federation Proc, 11, 222 (1952).
21. Harman, J., J. Exp. Cell Res., 1, 382, 394 (1950).
22. Hogeboom, G. H., Schneider, W. C, and Pallade, G. E.,J. Biol. Chem., 172, 619 (1948).
23. Johnson, R. B., and Lardy, H. A.,J. Biol. Chem., 184, 235 (1950).
24. Kennedy, E. P., unpub.
25. Kennedy, E. P., and Barker, H. A., J. Biol. Chem., 191, 419 (1951).
26. Kennedy, E. P., and Lehninger, A. L., J. Biol. Chem., 172, 847 (1948).
27. Kennedy, E. P., and Lehninger, A. L., J. Biol. Chem., 179, 957 (1949).
28. Kennedy, E. P., and Lehninger, A. L., J. Biol. Chem., 185, 275 (1950).
29. Kennedy, E. P., and Lehninger, A. L., J. Biol. Chem., 190, 361 (1951).
30. Knox, W. E., Noyce, B. N., and Auerbach, V. H., J. Biol. Chem., 176, 117 (1948).
31. Kornberg, A., and Pricer, W. E., Jr., /. Amer. Chem. Soc, 74, 1617 (1952).
32. Lardy, H., in Phosphorus Metabolism, I (W. D. McElroy & B. Glass, eds.), Johns Hopkins Press, Baltimore (1951).
33. Lehninger, A. L., unpub.
34. Lehninger, A. L., J. Biol. Chem., 154, 309 (1944).
35. Lehninger, A. L., J. Biol. Chem., 157, 363 (1945).
36. Lehninger, A. L., J. Biol. Chem., 161, 413 (1945).
37. Lehninger, A. L., J. Biol. Chem., 161, 437 (1945).
38. Lehninger, A. L, J. Biol. Chem., 162, 333 (1946).
39. Lehninger, A. L.,J. Biol. Chem., 164, 291 (1946).
40. Lehninger, A. L.,J. Biol. Chem., 165, 131 (1946).
41. Lehninger, A. L.,J. Biol. Chem., 190, 345 (1951).
42. Lehninger, A. L.,J. and Kennedy, E. P., J. Biol. Chem., 174, 883 (1948).
43. Lehninger, A. L., and Smith, S. W., J. Biol. Chem., 181, 415 (1949).
44. Leloir, L. F., and Munoz, J. M.,J. Biol. Chem., 153, 53 (1944).
45. Lipmann, F., Jones, M. E., Black, S., and Flynn, R. M., J. Amer. Chem. Soc, 74, 2384 (1952).
46. Lipmann, F-, and Perlmann, G. E., Arch. Biochem., 1, 41 (1942).
47. Lorber, V., Cook, M., and Meyer, J,J. Biol. Chem., 181, 475 (1949).
48. Lynen, F., Reichert, E., and Rueff, L., Ann. Chem., 574, 1 (1951).
49. Medes, G., Weinhouse, S., and Floyd, N. F, J. Biol. Chem., 157, 35 (1945).
50. Munoz, J. M., and Leloir, L. F, J. Biol. Chem., 147, 355 (1943).
51. Ochoa, S., Stern, J. R., and Schneider, M. C,J. Biol. Chem., 193, 691 (1952).
52. Potter, V. R., J. Biol. Chem., 163, 437 (1946).
53. Rittenberg, D., and Bloch, K, J. Biol. Chem., 154, 311 (1944); 160, 417 (1945).
54. Schneider, W. C,J. Biol. Chem., 176, 259 (1948).
55. Stadie, W. C, Physiol. Rev., 25, 395 (1945).
56. Stadtman, E. R., and Barker, H. A.,J. Biol. Chem., 180, 1085, 1095, 1117, 1169 (1949).
57. Stadtman, E. R., and Barker, H. A., J. Biol. Chem., 184, 769 (1950).
58. Stadtman, E. R., Doudoroff, M., and Lipmann, F., J. Biol. Chem., 191, 377 (1951).
59. Stadtman, E. R., Novelli, G. D., and Lipmann, F., J. Biol. Chem., 191, 365 (1951).
60. Stern, J. R., and Ochoa, S., /. Biol. Chem., 191, 161 (1951).
61. Stern, J. R., Shapiro, B., Stadtman, E. R., Ochoa, S., J. Biol. Chem., 193, 703 (1951).
62. Weinhouse, S., Cancer Res., 11, 585 (1951).
63. Weinhouse, S., Medes, G., and Floyd, N. F., J. Biol. Chem., 155, 143 (1944).
64. Weinhouse, S., Millington, R. H., and Volk, M. E., J. Biol. Chem., 185, 191 (1950).
65. Wieland, H., and Rosenthal, C, Ann. Chem., 554, 241 (1943).
66. Zabin, I.,J. Biol. Chem., 189, 355 (1951).
Continue to:
My Books

