Mechanisms Of Hormone Action. Part 12
Description
This section is from the book "The Scientific Contributions Of The Ben May Laboratory For Cancer Research", by The University of Chicago. Also available from Amazon: The Scientific Contributions Of The Ben May Laboratory For Cancer Research.
Mechanisms Of Hormone Action. Part 12
2. Model Experiments With Peroxidases
Peroxidases catalyze the oxidation of hydrogen donors upon the addition of hydrogen peroxide. But under certain circumstances, peroxidases will function as aerobic oxidases. For example, dihydroxyfumaric acid (18, 119) and phenylacetaldehyde (69) are oxidized by peroxidases in the presence of oxygen and low levels of Mn++ ions and in the absence of added hydrogen peroxide. Akazawa and Conn (2) showed that certain monophenols mediate the aerobic oxidation of many hydrogen or electron donors by a horseradish peroxidase-Mn++ system, e.g., reduced pyridine nucleotides, ferrocyto-chrome c, dicarboxylic acids, indoleacetic acid, and reduced glutathione. These oxidations were inhibited by cyanide, and were not catalyzed by a variety of other heme proteins. Catalase proved to be a powerful inhibitor, despite the fact that hydrogen peroxide did not have to be added for the oxidations to proceed, and did not accumulate during the course of the reaction.
Although they are widely distributed in the plant kingdom, it is often surmised that peroxidases play a relatively unimportant role in the metabolism of animal tissues. The weak peroxidase activity of hemoglobin and other heme proteins, and the oxidation of some peroxidase substrates by the cytochrome c-cytochrome oxidase system, casts doubt on the validity of many early reports on the presence of peroxidases in animal tissues. In 1934, Bancroft and Elliott (7) provided strong evidence for peroxidases in lung and spleen. Agner has isolated a green peroxidase (verdoperoxidase) from human leucocytes (1). More recently, a series of careful experiments were carried out by Stotz and his associates (94), in which every effort was taken to minimize interference by nonspecific hemes and heme proteins in the assay for peroxidases in crude tissue extracts. They found that rat tissues with a high rate of cell renewal (uterus, small intestine, spleen, lung, and Walker 256 carcinoma) exhibited high levels of peroxidase, but relatively weak cytochrome oxidase activity. Conversely, tissues with a very active cytochrome oxidase (liver, muscle, kidney, and brain) were poor in peroxidase. Previous experiments by these workers (80) showed that the peroxidase levels of rat uterus were extremely sensitive to the action of estrogens in vivo. Enzyme activity was barely perceptible in homogenates of the uteri of spayed animals, but increased more than 200-fold after the injection of either estradiol-17β or diethylstilbestrol. This action of both natural and synthetic estrogens was apparent within 24 hours after their administration. In terms of its ability to oxidize various donors in the presence of hydrogen peroxide, uterine peroxidase resembles lactoperoxidase, but is quite different from verdoperoxidase, yeast cytochrome c peroxidase, and the peroxidase of horseradish and carrot (84).
We have examined the possible carrier function of phenolic estrogens in the aerobic, peroxidase-catalyzed oxidation of many donors in the absence of added hydrogen peroxide. It is of interest that Huszak, in the laboratory of Albert Szent-Gydrgyi (57), provided evidence for a catalytic role of certain flavonoids (which are chemically related to plant estrogens like genistein) in peroxidase-catalyzed reactions; but in these experiments, hydrogen peroxide was added as the oxidant.
At pH 7.4, purified preparations of either horseradish peroxidase or lactoperoxidase barely oxidize DPNH or TPNH in the presence of trace levels (10-5M) of Mn++. Many estrogenic phenols stimulate the oxidation of these nucleotides under such conditions (138, 139) as Akazawa and Conn (2) have described for a number of simple phenols. In these systems, the oxidation of DPNH is inhibited by cyanide, and by catalase. In marked contrast to the phenolase-catalyzed reactions described above, no induction period was observed with estradiol and many other monophenolic estrogens. Also epinephrine, norepinephrine and 3-hydroxytyramine (which function as carriers in the phenolase-catalyzed oxidation of DPNH) did not mediate the oxidation of DPNH by the horseradish peroxidase-Mn+ + system, and inhibited the action of estradiol-17β therein. The optimum pH for the oxidation of DPNH varied with different estrogenic phenols, but in general lay between pH 8.0 and 8.5. Manometric experiments showed that the estradiol-mediated oxidation of DPNH proceeded to completion with the consumption of one atom of oxygen per mole of nucleotide that was oxidized. Hydrogen peroxide was not required for this reaction and was very inhibitory if present at a concentration greater than 10-4 M. Estradiol-17β also increased the rate of oxidation of ferrocytochrome c by horseradish peroxidase in the absence of hydrogen peroxide, a reaction which did not exhibit a marked requirement for Mn++. For the oxidation of both DPNH and ferrocytochrome c at pH 7.4, 50% of the maximal carrier activity of estradiol-17β was observed when its final concentration was 8 X 10-6 M.
Akazawa and Conn (2) have suggested the following mechanism for the phenol-stimulated oxidation of DPNH by peroxidases. A ternary complex of peroxidase, hydrogen peroxide, and Mn++ catalyzes the oxidation of the phenol (ROH) to an oxidized product (RO.) and hydrogen peroxide. This oxidized product is then reduced by the pyridine nucleotide. The hydrogen peroxide generated in the first reaction may oxidize the phenol, in a typical peroxidatic reaction, to an oxidized product. Thus, the oxidation of DPNH would proceed according to the following equations:
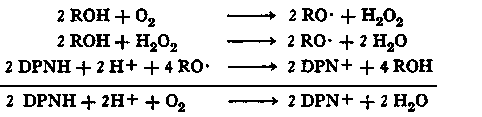
This reaction mechanism accounts for the stoichiometry of DPNH oxidation and oxygen utilization, and also for the fact that Akazawa and Conn (2) could not detect any accumulation of hydrogen peroxide during the course of the reaction. The chemical nature of the oxidized phenol (RO.) is a matter of conjecture, but it is not unreasonable to postulate that, in the case of monophenols, the oxidation products are phenoxy radicals, which might be stabilized by the enzyme as a peroxidase-phenoxy radical complex. Akazawa and Conn (2) point out that the oxidation of reduced pyridine nucleotides by such a peroxidase-phenoxy radical complex would be analogous to the oxidation of various donors by a peroxidase-hydrogen peroxide complex (19). In the case of monophenolic estrogens such as estradiol-17 6, the formation of phenoxy radical products would provide a simple explanation for the mediation of the oxidation of DPNH by the horseradish per-oxidase-Mn++ system and would be in accord with our finding that, in experiments of short duration, estradiol-17β can be recovered unchanged from this oxidase system. In the case of hexestrol and dienestrol, which are active carriers in this system, it is possible that either mono- or diradical oxidation products are the hydrogen transporting species. With diethylstilbestrol, a marked lag period is observed for the oxidation of DPNH. Preliminary experiments (140) suggest that diethylstilbestrol may undergo oxidation to isodienestrol (122) which may, in turn, transport hydrogen by oxidation to a free radical form.
3. Uterine Peroxidase
Hollander and Stephens (51) have described an enzyme system in rat uterus for the oxidation of reduced pyridine nucleotides, which is activated by low concentrations of Mn++ and certain simple monophenols such as 2,4-dichlorophenol. DPNH was oxidized at the same rate as TPNH, and one atom of oxygen was consumed per mole of the nucleotide which was oxidized. The oxidations proceeded without the addition of hydrogen peroxide, although a lag period was observed with some enzyme preparations which could be shortened by the addition of very low levels of hydrogen peroxide (an excess of which inhibited the oxidation of DPNH completely).
Continue to:
My Books

